Sie haben das Marfan-Syndrom oder es besteht der Verdacht darauf?
Diese Information soll Ihnen helfen, sich einen ersten Überblick über dieses seltene Krankheitsbild zu verschaffen. Wenn Sie mehr über die Erkrankung wissen, können Sie ihr besser begegnen.
Auf einen Blick
Erkrankung
Das Marfan-Syndrom ist eine seltene erbliche Erkrankung, die das Bindegewebe betrifft. Es gibt viele verschiedene Anzeichen wie: ein schmaler, langer Körper, überdehnbare Gelenke, Abreißen oder Verschieben der Augenlinse, Ausweitungen und Risse in Blutgefäßen. Diese können unterschiedlich stark ausgeprägt sein.
Behandlung
Behandlungsmöglichkeiten sind: Medikamente, Operationen, Physiotherapie. Wichtig: Bestimmte körperliche Belastungen sind zu vermeiden, etwa schweres Heben.
Die Erkrankung
Das Marfan-Syndrom ist eine erblich bedingte Erkrankung. Durch einen genetischen Fehler bildet sich das Bindegewebe nicht normal. Bindegewebe kommt fast überall im Körper vor. Betroffen sind daher viele verschiedene Organe und Körperstrukturen, wie Knochen und Gelenke, Augen oder Herz und Blutgefäße.
Nach Schätzungen sind etwa 1 bis 2 von 10 000 Menschen am Marfan-Syndrom erkrankt. Die Krankheit wird unabhängig vom Geschlecht vererbt. Männer und Frauen sind gleich häufig betroffen. Ist ein Elternteil erkrankt, besteht eine Wahrscheinlichkeit von 50 Prozent, dass auch das Kind erkrankt. Bei etwa jedem Vierten wird die Krankheit nicht vererbt, sondern die Erbinformation hat sich zufällig verändert. Das Marfan-Syndrom tritt dann in dieser Familie neu auf.
Anzeichen und Beschwerden
Es treten verschiedene Auffälligkeiten auf, die unterschiedlich ausgeprägt sein können: stark, schwach oder gar nicht. Viele Anzeichen sind im Kindesalter noch nicht vorhanden und entwickeln sich erst mit der Zeit. Folgende Krankheitszeichen können auftreten:
-
Herz und Gefäße: ausgeweitete Gefäße, vor allem die Hauptschlagader (Aorta) ist betroffen, Risse der Gefäße, Herzklappenfehler, entzündete Herzklappen, Herzschwäche, Herzversagen
-
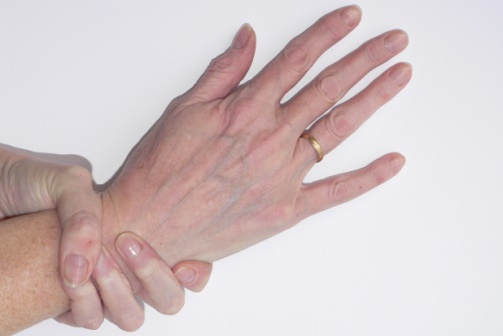
Knochen und Gelenke: lange, schmale Finger, Arme und Beine, schmaler Körperbau, Hochwuchs, verkrümmte Wirbelsäule, verformtes Brustbein, Platt-, Knick-, Senkfuß, dehnbare Gelenke, langer Schädel, Zahnfehlstellungen. Typisch ist, dass sich die Endglieder von Daumen und kleinem Finger komplett überlappen, wenn man das eigene Handgelenk umfasst (positives Handgelenkszeichen).
-
Augen: Kurzsichtigkeit, Erblinden, Ablösen der Netzhaut, Abreißen oder Verschieben der Augenlinse, Linsentrübung (grauer Star in jungen Jahren)
-
Lunge: Zusammenfall der Lunge
-
Haut: Dehnungsstreifen, Leistenbruch
Vor allem eine veränderte Aorta und krankhafte Herzklappen mindern die Lebenserwartung der Betroffenen. Ohne Behandlung werden sie im Mittel etwa 40 Jahre alt. Bei guter medizinischer Betreuung können sie heute genauso alt werden wie andere Menschen auch.
Untersuchungen
Der Arzt oder die Ärztin befragt Sie und achtet bei der körperlichen Untersuchung auf Anzeichen, die auf das Marfan-Syndrom hindeuten können. Sind die Ergebnisse eindeutig, so kann man sagen, ob ein Marfan-Syndrom vorliegt. Es könnte wichtig sein zu unterscheiden, ob Sie das Marfan-Syndrom oder eine ähnliche Erbkrankheit haben. Ein Gentest kann dann hilfreich sein.
Behandlungen
Das Marfan-Syndrom ist nicht heilbar, aber es gibt gute Behandlungsmöglichkeiten. Ein spezialärztliches Team aus den Fachgebieten Herzkrankheiten (Kardiologie), Orthopädie und Augenheilkunde soll Sie regelmäßig und dauerhaft betreuen.
Besonders ernst zu nehmen sind Folgen, die das Herz und die Gefäße betreffen. So besteht ein Risiko, dass sich die Aorta ausweitet. Wenn sie reißt, besteht Lebensgefahr. Möglicherweise können bestimmte Blutdruck senkende Medikamente, sogenannte Beta-Blocker, dem entgegenwirken und das Überleben verbessern. Kommen Beta-Blocker nicht in Frage, sind als Blutdruck-Medikamente auch Sartane (Angiotensin-Rezeptor-Blocker) möglich.
Manchmal empfehlen die Fachleute zusätzlich eine vorbeugende Operation der Aorta. Dabei entfernt das Ärzteteam die ausgeweitete Gefäß-Stelle und ersetzt sie durch künstliches Gewebe. Ob eine solche Operation erfolgen sollte, hängt davon ab, wie stark das Gefäß ausgeweitet ist und wie schnell es sich verbreitert. Dabei spielt auch eine Rolle, welche Begleit-Erkrankungen Sie haben und wie das Marfan-Syndrom in Ihrer Familie verlaufen ist. Bei vorbeugender Operation überleben 98 bis 99 von 100 Betroffenen. Reißt die Aorta plötzlich, ist dies ein Notfall. Eine Notfall-Operation überleben etwa 80 von 100 Betroffenen.
An veränderten Herzklappen können sich leichter Bakterien festsetzen und zu Entzündungen des Herzens führen. In bestimmten Situationen, zum Beispiel vor zahnärztlichen Eingriffen, sollten Sie daher vorsorglich ein Antibiotikum erhalten.
Orthopädische Probleme lassen sich mit Physiotherapie behandeln. Operationen an Wirbelsäule, Füßen, Augen oder Lunge lindern manche Beschwerden.
Um das übermäßige Körperwachstum einzuschränken, kommen manchmal Hormone zum Einsatz.
Was Sie selbst tun können
-
Lassen Sie sich in einer Marfan-Sprechstunde von Fachleuten für Ihre Krankheit betreuen.
-
Gehen Sie regelmäßig zu den ärztlichen Kontroll-Untersuchungen. So kann man den Verlauf der Krankheit beurteilen und behandelbare Veränderungen rechtzeitig erkennen. Besonders wichtig sind häufige Blutdruck-Kontrollen und jährliche Herz-Untersuchungen.
-
Sollten sich Beschwerden auf einmal deutlich verschlechtern, holen Sie sofort ärztlichen Rat ein.
-
Bestimmte Anzeichen, wie Schmerzen im Brust- oder Bauchraum, Gefühl der Atemnot oder Ohnmacht, können auf einen Notfall hindeuten. Rufen Sie 112.
-
Bei Angehörigen ersten Grades ist die Wahrscheinlichkeit erhöht, dass sie selbst erkrankt sind. Eine gezielte Untersuchung kann hier Klarheit bringen.
-
Wenn Sie Fragen zur Vererbung der Krankheit haben, gehen Sie zu einem Zentrum für das Marfan-Syndrom mit ausführlicher Untersuchung und Beratung. Dort können Sie Ihr Blut oder das Ihres Kindes auf veränderte Erbanlagen untersuchen lassen.
-
Lassen Sie sich beraten, wenn Sie eine Schwangerschaft planen.
-
Bestimmte körperliche Belastungen, wie schweres Heben, Kraftsport, Wettkampf- oder Kontaktsport, erhöhen die Gefahr, dass die Aorta reißt. Fachleute raten daher davon ab.
-
Sie sind nicht allein mit Ihrer Erkrankung, es gibt Unterstützungs- und Beratungsangebote, die Ihnen den Alltag erleichtern können. Informieren Sie sich über Selbsthilfe-Organisationen und tauschen Sie Ihre Erfahrungen mit anderen Betroffenen aus.
In der Allianz Chronischer Seltener Erkrankungen (ACHSE) e. V. haben sich Patientenorganisationen zusammengeschlossen und sich auf gemeinsame Standards für eine unabhängige Selbsthilfearbeit geeinigt. Ansprechpersonen für Ihre Erkrankung finden Sie hier:
Internet www.achse-online.de
Telefon 030 3300708-0
E-Mail info@achse-online.de
Selbsthilfe-Organisation:
Unsere Gesundheitsinformationen können Sie kostenlos herunterladen, ausdrucken und verteilen. Es gibt auch die Möglichkeit, diese bei Anbietern von Print on Demand auf hochwertigem Papier und in beliebiger Auflage kostenpflichtig ausdrucken zu lassen – wie zum Beispiel dem DDZ.
Diese Information wurde vom ÄZQ im Rahmen eines kooperativen Projektes mit der Allianz Chronischer Seltener Erkrankungen (ACHSE) e. V erstellt. Der Inhalt beruht auf aktuellen wissenschaftlichen Forschungsergebnissen und Empfehlungen für Betroffene von Betroffenen.
Hier finden Sie Dokumente zur Methodik, alle Quellen der Kurzinformation "Marfan-Syndrom" sowie weiterführende Links.
Methodik
Verwendete Quellen
Fachliteratur – Nachrecherche (11/2020)
-
Al-Abcha A, Saleh Y, Mujer M, et al. Meta-analysis Examining the Usefulness of Angiotensin Receptor blockers for the Prevention of Aortic Root Dilation in Patients With the Marfan Syndrome. Am J Cardiol 2020; 128:101–6. DOI: 10.1016/j.amjcard.2020.04.034. www.ncbi.nlm.nih.gov/pubmed/32650901.
-
Baumgartner H, Backer J de, Babu-Narayan SV, et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur Heart J 2020. DOI: 10.1093/eurheartj/ehaa554. www.ncbi.nlm.nih.gov/pubmed/32860028.
-
Cardiac Society of Australia and New Zealand (CSANZ). Update on the diagnosis and management of inherited aortopathies, including Marfan syndrome. 2016 [cited: 2021-01-29]. www.csanz.edu.au/wp-content/uploads/2016/11/Inherited_aortopathies_inc_Marfan_Syndrome_ratified_25-Nov-2016.pdf.
-
Demolder A, Kodolitsch Y von, Muiño-Mosquera L, et al. Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics (Basel, Switzerland) 2020; 10(10). DOI: 10.3390/diagnostics10100751. www.ncbi.nlm.nih.gov/pubmed/32992882.
-
Elbadawi A, Omer MA, Elgendy IY, et al. Losartan for Preventing Aortic Root Dilatation in Patients with Marfan Syndrome: A Meta-Analysis of Randomized Trials. Cardiol Ther 2019; 8(2):365–72. DOI: 10.1007/s40119-019-00149-3. www.ncbi.nlm.nih.gov/pubmed/31606871.
-
Gao L, Chen L, Fan L, et al. The effect of losartan on progressive aortic dilatation in patients with Marfan's syndrome: A meta-analysis of prospective randomized clinical trials. Int J Cardiol 2016; 217:190–4. DOI: 10.1016/j.ijcard.2016.04.186. www.ncbi.nlm.nih.gov/pubmed/27187761.
-
Kang Y-N, Chi S-C, Wu M-H, et al. The effects of losartan versus beta-blockers on cardiovascular protection in marfan syndrome: A systematic review and meta-analysis. J Formos Med Assoc 2020; 119(1 Pt 1):182–90. DOI: 10.1016/j.jfma.2019.03.018. www.ncbi.nlm.nih.gov/pubmed/31003918.
-
Kiotsekoglou A, Moggridge JC, Child AH, et al. The role of advanced echocardiography and cardiovascular magnetic resonance in the assessment of myocardial function in Marfan syndrome-An update. Echocardiography 2017; 34(5):760–7. DOI: 10.1111/echo.13517. www.ncbi.nlm.nih.gov/pubmed/28317279.
-
Koo H-K, Lawrence KA, Musini VM. Beta-blockers for preventing aortic dissection in Marfan syndrome. Cochrane Database Syst Rev 2017; 11:CD011103. DOI: 10.1002/14651858.CD011103.pub2. www.ncbi.nlm.nih.gov/pubmed/29110304.
-
Li L, Yamani N, Al-Naimat S, et al. Role of losartan in prevention of aortic dilatation in Marfan syndrome: A systematic review and meta-analysis. Eur J Prev Cardiol 2020; 27(13):1447–50. DOI: 10.1177/2047487319861231. www.ncbi.nlm.nih.gov/pubmed/31260329.
-
Malik AH, Yandrapalli S, Pemmasani G, et al. Pharmacotherapeutics for prevention of aortic root enlargement in Marfan Syndrome - A network meta-analysis of randomized controlled trials. Eur J Prev Cardiol 2020; 27(19):2187–90. DOI: 10.1177/2047487319874907. www.ncbi.nlm.nih.gov/pubmed/31487999.
-
Nielsen C, Ratiu I, Esfandiarei M, et al. A Review of Psychosocial Factors of Marfan Syndrome: Adolescents, Adults, Families, and Providers. Journal of pediatric genetics 2019; 8(3):109–22. DOI: 10.1055/s-0039-1693663. www.ncbi.nlm.nih.gov/pubmed/31406616.
Weiterführende Links
Diese Auflistung ist eine Auswahl, sie wird fortlaufend ergänzt und ist nicht vollständig.
- www.orpha.net
- Für Patientinnen und Patienten mit Marfan-Syndrom gibt es eine besondere Form der ärztlichen Versorgung: die ambulante spezialfachärztliche Versorgung (kurz: ASV)
- Im ASV-Verzeichnis können Sie geeignete Behandlungsangebote in Ihrer Nähe finden: www.asv-servicestelle.de



Hinweise und Kommentare
Sie haben Hinweise und Kommentare zu unserem Internetangebot?